



VWF Rabbit pAb
-YN2038

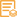


主要信息
Target
VWF
Host Species
Rabbit
Reactivity
Human, Mouse, Rat
Applications
IHC, IF
MW
309kD (Observed)
Conjugate/Modification
Unmodified

货号: YN2038
规格
价格
货期
数量
200μL
¥3,780.00
一个月
0
100μL
¥2,300.00
一个月
0
40μL
¥960.00
一个月
0
加入购物车


已收藏
收藏
详细信息
推荐稀释比
IHC 1:50-300; IF 1:50-200
Note:For IHC,wesuggest antigen retrieval with TE buffer pH 9.0 (Cat#YS0004)
Note:For IHC,wesuggest antigen retrieval with TE buffer pH 9.0 (Cat#YS0004)
组成
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
特异性
VWF Polyclonal Antibody detects endogenous levels of protein.
纯化工艺
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
储存
-15°C to -25°C/1 year(Do not lower than -25°C)
浓度
1 mg/ml
实测条带
309kD
修饰
Unmodified
克隆性
Polyclonal
同种型
IgG
相关产品
Secondary Antibodies
Goat Anti Mouse IgG(H+L) (HRP)
RS0001
预览→
Secondary Antibodies
Goat Anti Rabbit IgG(H+L) (HRP)
RS0002
预览→
Primary Antibodies
β-actin (PTR2364) Mouse mAb
YM3028
预览→
Primary Antibodies
GAPDH (PTR2304) Mouse mAb
YM3029
预览→
抗原&靶点信息
免疫原:
Synthesized peptide derived from part region of human protein AA range: 911-960
展开内容
特异性:
VWF Polyclonal Antibody detects endogenous levels of protein.
展开内容
基因名称:
VWF F8VWF
展开内容
蛋白名称:
von Willebrand factor (vWF) [Cleaved into: von Willebrand antigen 2 (von Willebrand antigen II)]
展开内容
背景:
This gene encodes a glycoprotein involved in hemostasis. The encoded preproprotein is proteolytically processed following assembly into large multimeric complexes. These complexes function in the adhesion of platelets to sites of vascular injury and the transport of various proteins in the blood. Mutations in this gene result in von Willebrand disease, an inherited bleeding disorder. An unprocessed pseudogene has been found on chromosome 22. [provided by RefSeq, Oct 2015],
展开内容
功能:
Disease:Defects in VWF are associated with various forms of von Willebrand disease (VWD) [MIM:193400, 277480]. VWD is characterized by frequent bleeding (gingival, minor skin quantitative lacerations, menorrhagia, etc.). Type I VWD is associated with a deficiency of VWF; type II by normal to decreased plasma level of VWF; type III by a virtual absence of VWF. There are subtypes (A to H) of type II VWD; for example: type IIA is characterized by the absence of VWF high molecular weight multimers in plasma.,Domain:The von Willebrand antigen 2 is required for multimerization of vWF and for its targeting to storage granules.,Function:Important in the maintenance of hemostasis, it promotes adhesion of platelets to the sites of vascular injury by forming a molecular bridge between sub-endothelial collagen matrix and platelet-surface receptor complex GPIb-IX-V. Also acts as a chaperone for coagulation factor VIII, delivering it to the site of injury, stabilizing its heterodimeric structure and protecting it from premature clearance from plasma.,online information:von Willebrand factor (vWF) mutation db,online information:Von Willebrand factor entry,PTM:All cysteine residues are involved in intrachain or interchain disulfide bonds.,similarity:Contains 1 CTCK (C-terminal cystine knot-like) domain.,similarity:Contains 3 VWFA domains.,similarity:Contains 3 VWFC domains.,similarity:Contains 4 TIL (trypsin inhibitory-like) domains.,similarity:Contains 4 VWFD domains.,subcellular location:Localized to storage granules.,subunit:Multimeric. Interacts with F8.,tissue specificity:Plasma.,
展开内容
细胞定位:
Secreted . Secreted, extracellular space, extracellular matrix . Localized to storage granules.
展开内容
组织表达:
展开内容
研究领域:
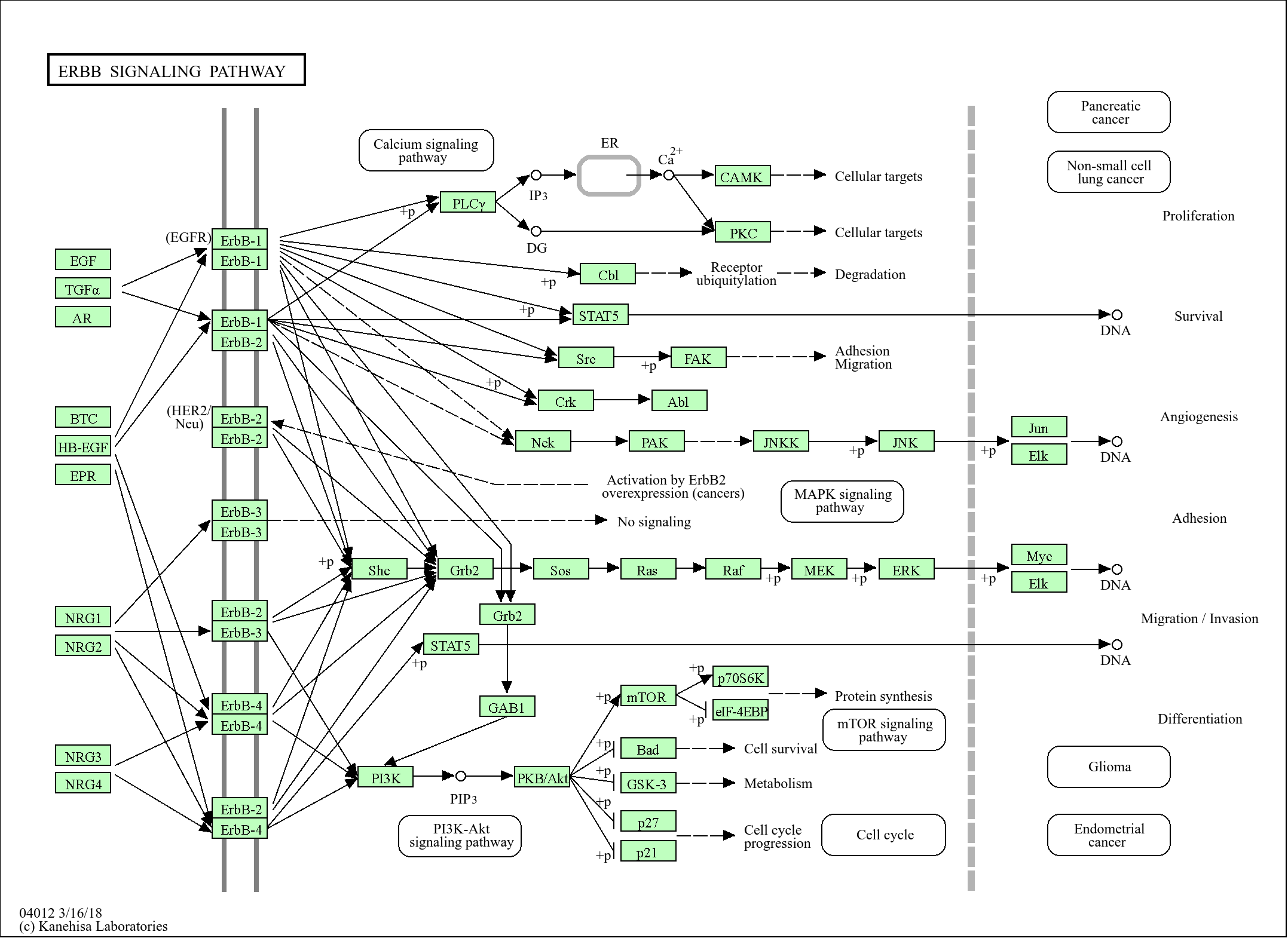
>>PI3K-Akt signaling pathway ;
>>Focal adhesion ;
>>ECM-receptor interaction ;
>>Complement and coagulation cascades ;
>>Platelet activation ;
>>Neutrophil extracellular trap formation ;
>>Human papillomavirus infection ;
>>Coronavirus disease - COVID-19
>>Focal adhesion ;
>>ECM-receptor interaction ;
>>Complement and coagulation cascades ;
>>Platelet activation ;
>>Neutrophil extracellular trap formation ;
>>Human papillomavirus infection ;
>>Coronavirus disease - COVID-19
展开内容
信号通路
Cellular Processes >> Cellular community - eukaryotes >> Focal adhesion
Organismal Systems >> Immune system >> Complement and coagulation cascades
Organismal Systems >> Immune system >> Platelet activation
Organismal Systems >> Immune system >> Neutrophil extracellular trap formation
Environmental Information Processing >> Signal transduction >> PI3K-Akt signaling pathway
Environmental Information Processing >> Signaling molecules and interaction >> ECM-receptor interaction
文献引用({{totalcount}})
货号: YN2038
规格
价格
货期
数量
200μL
¥3,780.00
一个月
0
100μL
¥2,300.00
一个月
0
40μL
¥960.00
一个月
0
加入购物车
已收藏
收藏
Recently Viewed Products
Clear all







Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
主要信息
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
产品 {{index}}/{{pcount}}
上一个产品
下一个产品
{{pvTitle}}
滚轮缩放图片
{{pvDescr}}