



KCNQ1 Rabbit pAb
-YN1086



主要信息
Target
KCNQ1
Host Species
Rabbit
Reactivity
Human, Mouse, Rat
Applications
WB, ELISA
MW
74kD (Observed)
Conjugate/Modification
Unmodified

货号: YN1086
规格
价格
货期
数量
200μL
¥3,780.00
现货
0
100μL
¥2,300.00
现货
0
40μL
¥960.00
现货
0
加入购物车


已收藏
收藏
详细信息
推荐稀释比
WB 1:500-2000; ELISA 1:5000-20000
组成
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
特异性
KCNQ1 Polyclonal Antibody detects endogenous levels of protein.
纯化工艺
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
储存
-15°C to -25°C/1 year(Do not lower than -25°C)
浓度
1 mg/ml
实测条带
74kD
修饰
Unmodified
克隆性
Polyclonal
同种型
IgG
相关产品
Secondary Antibodies
Goat Anti Mouse IgG(H+L) (HRP)
RS0001
预览→
Secondary Antibodies
Goat Anti Rabbit IgG(H+L) (HRP)
RS0002
预览→
Primary Antibodies
β-actin (PTR2364) Mouse mAb
YM3028
预览→
Primary Antibodies
GAPDH (PTR2304) Mouse mAb
YM3029
预览→
抗原&靶点信息
免疫原:
Synthesized peptide derived from human protein . at AA range: 350-430
展开内容
特异性:
KCNQ1 Polyclonal Antibody detects endogenous levels of protein.
展开内容
基因名称:
KCNQ1 KCNA8 KCNA9 KVLQT1
展开内容
蛋白名称:
Potassium voltage-gated channel subfamily KQT member 1 (IKs producing slow voltage-gated potassium channel subunit alpha KvLQT1) (KQT-like 1) (Voltage-gated potassium channel subunit Kv7.1)
展开内容
背景:
This gene encodes a voltage-gated potassium channel required for repolarization phase of the cardiac action potential. This protein can form heteromultimers with two other potassium channel proteins, KCNE1 and KCNE3. Mutations in this gene are associated with hereditary long QT syndrome 1 (also known as Romano-Ward syndrome), Jervell and Lange-Nielsen syndrome, and familial atrial fibrillation. This gene exhibits tissue-specific imprinting, with preferential expression from the maternal allele in some tissues, and biallelic expression in others. This gene is located in a region of chromosome 11 amongst other imprinted genes that are associated with Beckwith-Wiedemann syndrome (BWS), and itself has been shown to be disrupted by chromosomal rearrangements in patients with BWS. Alternatively spliced transcript variants have been found for this gene. [provided by RefSeq,
展开内容
功能:
Alternative products:Additional isoforms seem to exist,Disease:Defects in KCNQ1 are the cause of atrial fibrillation type 3 (ATFB3) [MIM:607554]. Atrial fibrillation is a common disorder of cardiac rhythm that is hereditary in a small subgroup of patients. It is characterized by disorganized atrial electrical activity, progressive deterioration of atrial electromechanical function and ineffective pumping of blood into the ventricles. It can be associated with palpitations, syncope, thromboembolic stroke, and congestive heart failure.,Disease:Defects in KCNQ1 are the cause of Jervell and Lange-Nielsen syndrome type 1 (JLNS1) [MIM:220400]. JLNS1 is an autosomal recessive disorder characterized by congenital deafness, prolongation of the QT interval, syncopal attacks due to ventricular arrhythmias, and a high risk of sudden death.,Disease:Defects in KCNQ1 are the cause of long QT syndrome type 1 (LQT1) [MIM:192500]; also known as Romano-Ward syndrome (RWS). Long QT syndromes are heart disorders characterized by a prolonged QT interval on the ECG and polymorphic ventricular arrhythmias. They cause syncope and sudden death in response to exercise or emotional stress. LQT1 inheritance is an autosomal dominant.,Disease:Defects in KCNQ1 are the cause of short QT syndrome type 2 (SQT2) [MIM:609621]. Short QT syndromes are heart disorders characterized by idiopathic persistently and uniformly short QT interval on ECG in the absence of structural heart disease in affected individuals. They cause syncope and sudden death.,Domain:The segment S4 is probably the voltage-sensor and is characterized by a series of positively charged amino acids at every third position.,Function:Probably important in cardiac repolarization. Associates with KCNE1 (MinK) to form the I(Ks) cardiac potassium current. Elicits a rapidly activating, potassium-selective outward current. Muscarinic agonist oxotremorine-M strongly suppresses KCNQ1/KCNE1 current in CHO cells in which cloned KCNQ1/KCNE1 channels were coexpressed with M1 muscarinic receptors. May associate also with KCNE3 (MiRP2) to form the potassium channel that is important for cyclic AMP-stimulated intestinal secretion of chloride ions, which is reduced in cystic fibrosis and pathologically stimulated in cholera and other forms of secretory diarrhea.,miscellaneous:Mutagenesis experiments were carried out by expressing in Xenopus oocytes or COS-7 cells KCNQ1 mutants either individually (homomultimers) or in combination with both wild-type KCNQ1 (mut/wt homomultimers) and minK (heteromultimers).,online information:Congenital long QT syndrome website,online information:KCNQ1 mutations page,online information:KvLQT1 entry,similarity:Belongs to the potassium channel family. KQT subfamily.,subunit:Heterotetramer with KCNE1 (MinK) or KCNE3 (MiRP2). Interacts with CALM.,tissue specificity:Abundantly expressed in heart, pancreas, prostate, kidney, small intestine and peripheral blood leukocytes. Less abundant in placenta, lung, spleen, colon, thymus, testis and ovaries.,
展开内容
细胞定位:
Cell membrane ; Multi-pass membrane protein . Cytoplasmic vesicle membrane . Early endosome . Membrane raft . Endoplasmic reticulum . Basolateral cell membrane . Colocalized with KCNE3 at the plasma membrane (PubMed:10646604). Upon 17beta-oestradiol treatment, colocalizes with RAB5A at early endosome (PubMed:23529131). Heterotetramer with KCNQ5 is highly retained at the endoplasmic reticulum and is localized outside of lipid raft microdomains (PubMed:24855057). During the early stages of epithelial cell polarization induced by the calcium switch it removed from plasma membrane to the endoplasmic reticulum where it retained and it is redistributed to the basolateral cell surface in a PI3K-dependent manner at a later stage (PubMed:21228319). .
展开内容
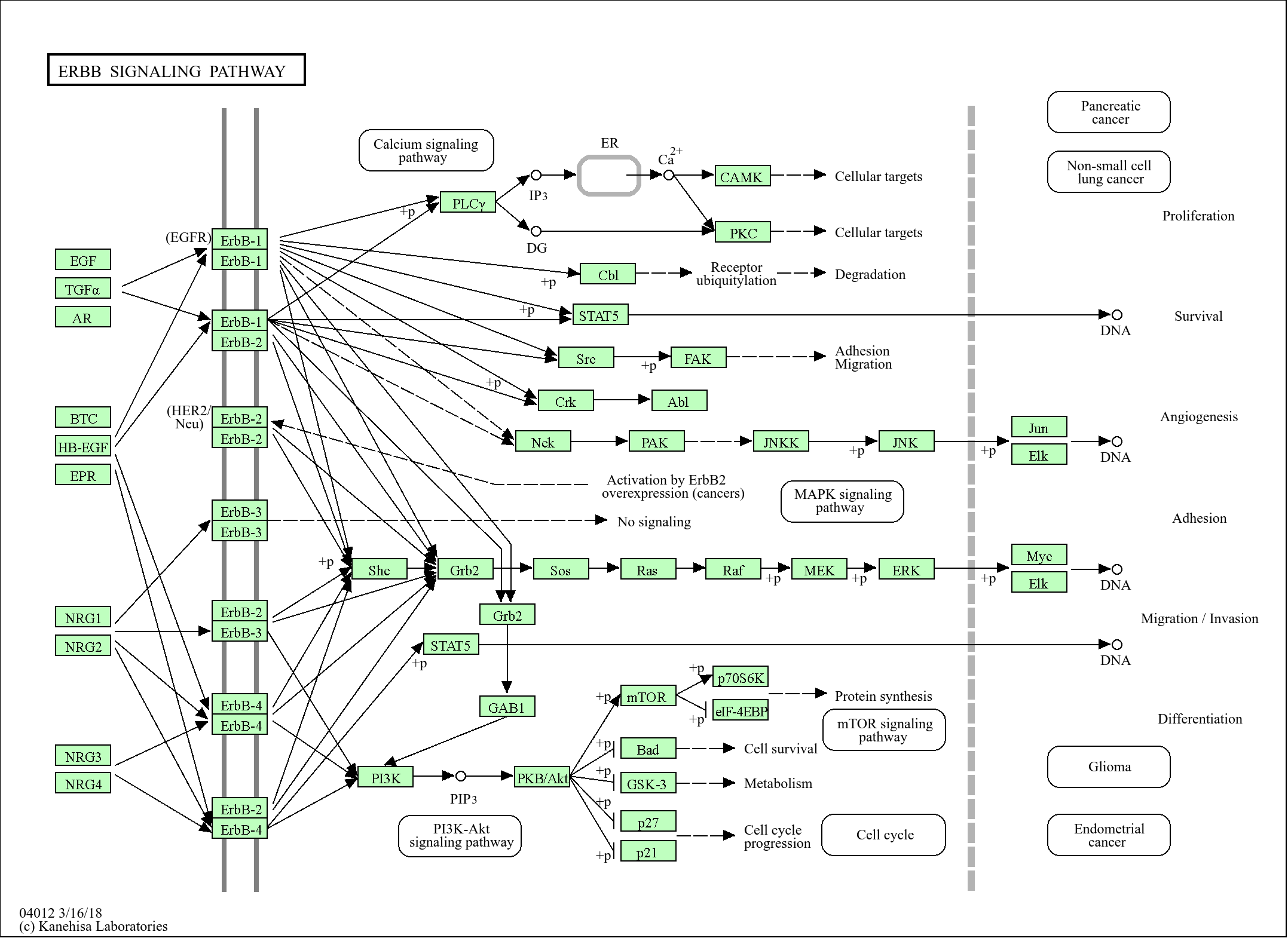
研究领域:
>>Adrenergic signaling in cardiomyocytes ;
>>Cholinergic synapse ;
>>Gastric acid secretion ;
>>Pancreatic secretion ;
>>Protein digestion and absorption ;
>>Vibrio cholerae infection
>>Cholinergic synapse ;
>>Gastric acid secretion ;
>>Pancreatic secretion ;
>>Protein digestion and absorption ;
>>Vibrio cholerae infection
展开内容
信号通路
文献引用({{totalcount}})
货号: YN1086
规格
价格
货期
数量
200μL
¥3,780.00
现货
0
100μL
¥2,300.00
现货
0
40μL
¥960.00
现货
0
加入购物车
已收藏
收藏
Recently Viewed Products
Clear all







Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
主要信息
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
产品 {{index}}/{{pcount}}
上一个产品
下一个产品
{{pvTitle}}
滚轮缩放图片
{{pvDescr}}