



MYH2 Rabbit pAb
-YN0898




主要信息
Target
MYH2
Host Species
Rabbit
Reactivity
Human
Applications
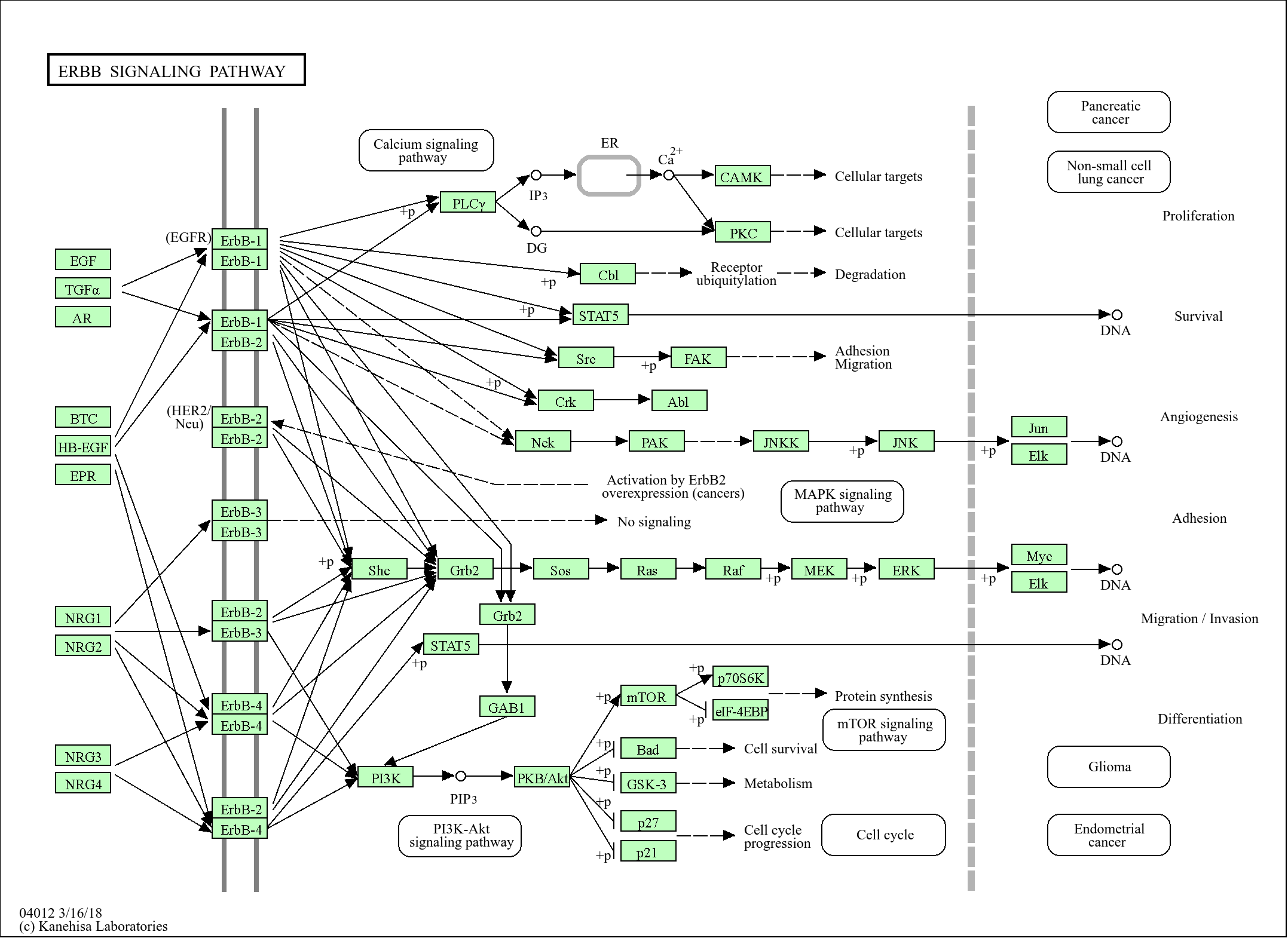
WB, ELISA
MW
213kD (Observed)
Conjugate/Modification
Unmodified

货号: YN0898
规格
价格
货期
数量
200μL
¥3,780.00
现货
0
100μL
¥2,300.00
现货
0
40μL
¥960.00
现货
0
加入购物车


已收藏
收藏
详细信息
推荐稀释比
WB 1:500-2000; ELISA 1:5000-20000
组成
Liquid in PBS containing 50% glycerol, and 0.02% sodium azide.
特异性
MYH2 Polyclonal Antibody detects endogenous levels of protein.
纯化工艺
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
储存
-15°C to -25°C/1 year(Do not lower than -25°C)
浓度
1 mg/ml
实测条带
213kD
修饰
Unmodified
克隆性
Polyclonal
同种型
IgG
相关产品
Secondary Antibodies
Goat Anti Mouse IgG(H+L) (HRP)
RS0001
预览→
Secondary Antibodies
Goat Anti Rabbit IgG(H+L) (HRP)
RS0002
预览→
Primary Antibodies
β-actin (PTR2364) Mouse mAb
YM3028
预览→
Primary Antibodies
GAPDH (PTR2304) Mouse mAb
YM3029
预览→
抗原&靶点信息
免疫原:
Synthesized peptide derived from human protein . at AA range: 760-840
展开内容
特异性:
MYH2 Polyclonal Antibody detects endogenous levels of protein.
展开内容
基因名称:
MYH2 MYHSA2
展开内容
蛋白名称:
Myosin-2 (Myosin heavy chain 2) (Myosin heavy chain 2a) (MyHC-2a) (Myosin heavy chain IIa) (MyHC-IIa) (Myosin heavy chain, skeletal muscle, adult 2)
展开内容
背景:
Myosins are actin-based motor proteins that function in the generation of mechanical force in eukaryotic cells. Muscle myosins are heterohexamers composed of 2 myosin heavy chains and 2 pairs of nonidentical myosin light chains. This gene encodes a member of the class II or conventional myosin heavy chains, and functions in skeletal muscle contraction. This gene is found in a cluster of myosin heavy chain genes on chromosome 17. A mutation in this gene results in inclusion body myopathy-3. Multiple alternatively spliced variants, encoding the same protein, have been identified. [provided by RefSeq, Sep 2009],
展开内容
功能:
Disease:Defects in MYH2 are the cause of inclusion body myopathy type 3 (IBM3) [MIM:605637]. Hereditary inclusion body myopathies constitute a group of neuromuscular disorders characterized by slowly progressive distal and proximal weakness and a typical muscle pathology including rimmed vacuoles and filamentous inclusions. IBM3 is a variant of hereditary inclusion body myopathies and is characterized by autosomal dominant myopathy with joint contracture, ophthalmoplegia and rimmed vacuoles. Morphological analysis of muscle biopsies from patients indicate that the type 2A fibers frequently were abnormal, whereas other fiber types appeared normal.,Domain:The rodlike tail sequence is highly repetitive, showing cycles of a 28-residue repeat pattern composed of 4 heptapeptides, characteristic for alpha-helical coiled coils.,Function:Muscle contraction. Required for cytoskeleton organization.,miscellaneous:Each myosin heavy chain can be split into 1 light meromyosin (LMM) and 1 heavy meromyosin (HMM). It can later be split further into 2 globular subfragments (S1) and 1 rod-shaped subfragment (S2).,similarity:Contains 1 IQ domain.,similarity:Contains 1 myosin head-like domain.,subcellular location:Thick filaments of the myofibrils.,subunit:Muscle myosin is a hexameric protein that consists of 2 heavy chain subunits (MHC), 2 alkali light chain subunits (MLC) and 2 regulatory light chain subunits (MLC-2).,
展开内容
细胞定位:
Cytoplasm, myofibril. Thick filaments of the myofibrils.
展开内容
组织表达:
Cerebellum,Muscle pool- 2 tissues- cardiac and skeletal muscle.,Skeletal muscle,
展开内容
文献引用({{totalcount}})
货号: YN0898
规格
价格
货期
数量
200μL
¥3,780.00
现货
0
100μL
¥2,300.00
现货
0
40μL
¥960.00
现货
0
加入购物车
已收藏
收藏
Recently Viewed Products
Clear all







Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
主要信息
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
产品 {{index}}/{{pcount}}
上一个产品
下一个产品
{{pvTitle}}
滚轮缩放图片
{{pvDescr}}