





Perforin (ABT236R) Rabbit mAb
-YM7190





主要信息
Target
Perforin
Host Species
Rabbit
Reactivity
Human
Applications
IHC, WB, ELISA
MW
61kD (Calculated)
Conjugate/Modification
Unmodified
货号: YM7190
规格
价格
货期
数量
200μL
¥3,780.00
现货
0
100μL
¥2,300.00
现货
0
40μL
¥960.00
现货
0
加入购物车


已收藏
收藏
详细信息
推荐稀释比
IHC 1:100-500; WB 1:500-1000; ELISA 1:5000-20000
组成
PBS, 50% glycerol, 0.05% Proclin 300, 0.05%BSA
特异性
This antibody detects endogenous levels of Perforin
纯化工艺
Recombinant Expression and Affinity purified
储存
-15°C to -25°C/1 year (Do not lower than -25°C)
理论分子量
61kD
修饰
Unmodified
克隆性
Monoclonal
克隆号
ABT236R
同种型
IgG1, Kappa
相关产品
Secondary Antibodies
Goat Anti Mouse IgG(H+L) (HRP)
RS0001
预览→
Secondary Antibodies
Goat Anti Rabbit IgG(H+L) (HRP)
RS0002
预览→
Primary Antibodies
β-actin (PTR2364) Mouse mAb
YM3028
预览→
Primary Antibodies
GAPDH (PTR2304) Mouse mAb
YM3029
预览→
抗原&靶点信息
免疫原:
Synthesized peptide derived from human Perforin AA range:1-100
展开内容
特异性:
This antibody detects endogenous levels of Perforin
展开内容
基因名称:
PRF1
展开内容
蛋白名称:
Perforin-1
展开内容
别名:
Cytolysin ;
FLH2 ;
HPLH2 ;
Lymphocyte pore-forming protein ;
P1 ;
PERF_HUMAN ;
perforin 1 ;
pore forming protein ;
Perforin 1 ;
Perforin-1 ;
PFP ;
PGFL ;
PIGF ;
PIGF-2 ;
PLGF ;
Pore forming protein ;
prf1 ;
SHGC-10760
FLH2 ;
HPLH2 ;
Lymphocyte pore-forming protein ;
P1 ;
PERF_HUMAN ;
perforin 1 ;
pore forming protein ;
Perforin 1 ;
Perforin-1 ;
PFP ;
PGFL ;
PIGF ;
PIGF-2 ;
PLGF ;
Pore forming protein ;
prf1 ;
SHGC-10760
展开内容
背景:
The protein encoded by this gene has structural and functional similarities to complement component 9 (C9) . Like C9 , this protein creates transmembrane tubules and is capable of lysing non-specifically a variety of target cells. This protein is one of the main cytolytic proteins of cytolytic granules , and it is known to be a key effector molecule for T-cell- and natural killer-cell-mediated cytolysis. Defects in this gene cause familial hemophagocytic lymphohistiocytosis type 2 (HPLH2) , a rare and lethal autosomal recessive disorder of early childhood. Alternative splicing results in multiple transcript variants encoding the same protein. [provided by RefSeq , Jul 2008] ,
展开内容
功能:
Disease:Defects in PRF1 are the cause of familial hemophagocytic lymphohistiocytosis type 2 (FHL2) [MIM:603553]; also known as HPLH2. Familial hemophagocytic lymphohistiocytosis (FHL) is a genetically heterogeneous , rare autosomal recessive disorder. It is characterized by immune dysregulation with hypercytokinemia and defective natural killer cell function. The clinical features of the disease include fever , hepatosplenomegaly , cytopenia , hypertriglyceridemia , hypofibrinogenemia , and neurological abnormalities ranging from irritability and hypotonia to seizures , cranial nerve deficits , and ataxia. Hemophagocytosis is a prominent feature of the disease , and a non-malignant infiltration of macrophages and activated T lymphocytes in lymph nodes , spleen , and other organs is also found. ,Function:In the presence of calcium , perforin polymerizes into transmembrane tubules and is capable of lysing non-specifically a variety of target cells. ,induction:Repressed by contact with target cells. ,online information:Perforin entry ,online information:PRF1 mutation db ,similarity:Belongs to the complement C6/C7/C8/C9 family. ,similarity:Contains 1 C2 domain. ,similarity:Contains 1 EGF-like domain. ,similarity:Contains 1 MACPF domain. ,subcellular location:Cytoplasmic granules of cytolytic T-lymphocytes. ,
展开内容
细胞定位:
Cytoplasmic , Membranous
展开内容
组织表达:
展开内容
研究领域:
>>Apoptosis ;
>>Natural killer cell mediated cytotoxicity ;
>>Type I diabetes mellitus ;
>>Autoimmune thyroid disease ;
>>Allograft rejection ;
>>Graft-versus-host disease ;
>>Viral myocarditis
>>Natural killer cell mediated cytotoxicity ;
>>Type I diabetes mellitus ;
>>Autoimmune thyroid disease ;
>>Allograft rejection ;
>>Graft-versus-host disease ;
>>Viral myocarditis
展开内容
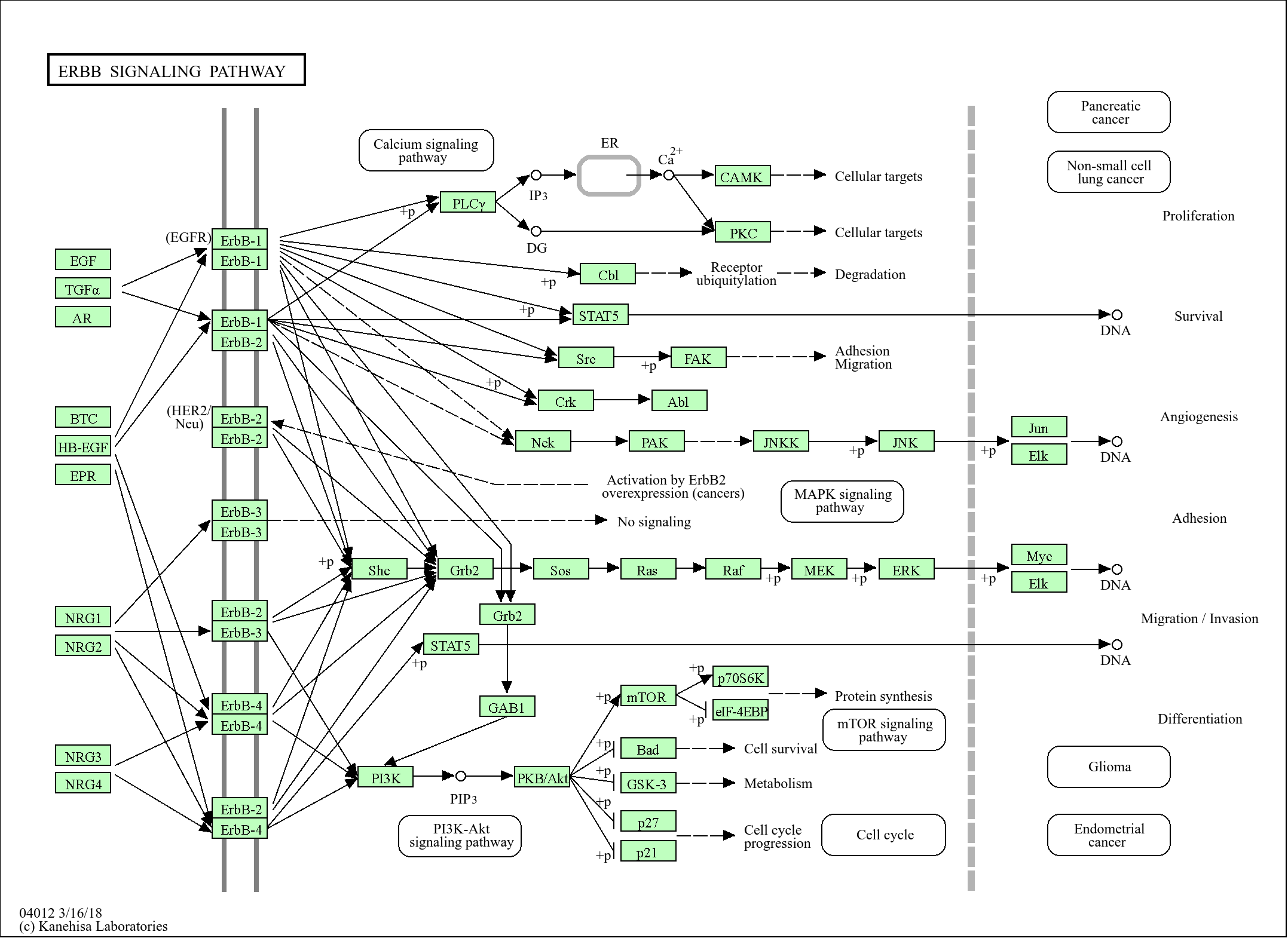
信号通路
Cellular Processes >> Cell growth and death >> Apoptosis
Organismal Systems >> Immune system >> Natural killer cell mediated cytotoxicity
Human Diseases >> Immune disease >> Autoimmune thyroid disease
Human Diseases >> Immune disease >> Allograft rejection
Human Diseases >> Immune disease >> Graft-versus-host disease
文献引用({{totalcount}})
货号: YM7190
规格
价格
货期
数量
200μL
¥3,780.00
现货
0
100μL
¥2,300.00
现货
0
40μL
¥960.00
现货
0
加入购物车
已收藏
收藏
Recently Viewed Products
Clear all







×

Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
主要信息
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
产品 {{index}}/{{pcount}}
上一个产品
下一个产品
{{pvTitle}}
滚轮缩放图片
{{pvDescr}}