
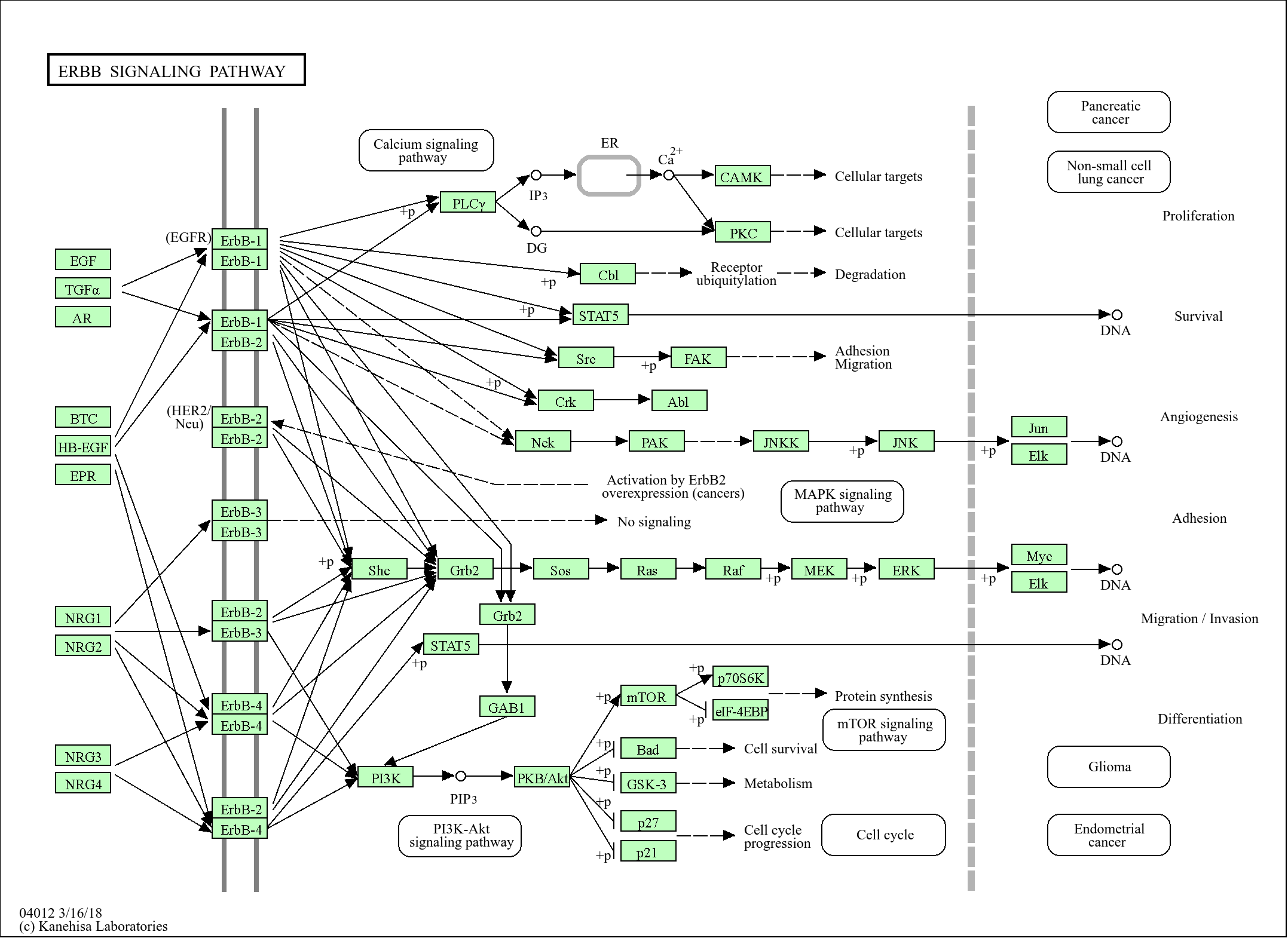




PKD2 (Phospho Ser812) Rabbit pAb
-YP1196


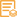


主要信息
Target
PKD2 Phospho Ser812
Host Species
Rabbit
Reactivity
Human, Mouse, Rat
Applications
WB, ELISA, IHC
MW
97kD (Calculated)
Conjugate/Modification
Phospho
货号: YP1196
规格
价格
货期
数量
200μL
¥4,680.00
现货
0
100μL
¥2,800.00
现货
0
50μL
¥1,500.00
现货
0
加入购物车


已收藏
收藏
详细信息
推荐稀释比
WB 1:500-2000; IHC 1:50-300; ELISA 1:2000-20000
组成
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
特异性
Phospho-PKD2 (S812) Polyclonal Antibody detects endogenous levels of PKD2S812.The name of modified sites may be influenced by many factors, such as species (the modified site was not originally found in human samples) and the change of protein sequence (the previous protein sequence is incomplete, and the protein sequence may be prolonged with the development of protein sequencing technology). When naming, we will use the "numbers" in historical reference to keep the sites consistent with the reports. The antibody binds to the following modification sequence (lowercase letters are modification sites):DDsEE
纯化工艺
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
储存
-15°C to -25°C/1 year (Do not lower than -25°C)
浓度
1 mg/ml
理论分子量
97kD
修饰
Phospho
克隆性
Polyclonal
同种型
IgG
相关产品
Primary Antibodies
PKD2 (Phospho Ser812) Rabbit pAb
YP1196
预览→
Secondary Antibodies
Goat Anti Mouse IgG(H+L) (HRP)
RS0001
预览→
Secondary Antibodies
Goat Anti Rabbit IgG(H+L) (HRP)
RS0002
预览→
Primary Antibodies
β-actin (PTR2364) Mouse mAb
YM3028
预览→
Primary Antibodies
GAPDH (PTR2304) Mouse mAb
YM3029
预览→
抗原&靶点信息
免疫原:
The antiserum was produced against synthesized peptide derived from human PKD2 around the phosphorylation site of Ser812. AA range:778-827
展开内容
特异性:
Phospho-PKD2 (S812) Polyclonal Antibody detects endogenous levels of PKD2S812.The name of modified sites may be influenced by many factors, such as species (the modified site was not originally found in human samples) and the change of protein sequence (the previous protein sequence is incomplete, and the protein sequence may be prolonged with the development of protein sequencing technology). When naming, we will use the "numbers" in historical reference to keep the sites consistent with the reports. The antibody binds to the following modification sequence (lowercase letters are modification sites):DDsEE
展开内容
基因名称:
PKD2
展开内容
蛋白名称:
Polycystin-2
展开内容
别名:
PKD2 ;
Polycystin-2 ;
Autosomal dominant polycystic kidney disease type II protein ;
Polycystic kidney disease 2 protein ;
Polycystwin ;
R48321
Polycystin-2 ;
Autosomal dominant polycystic kidney disease type II protein ;
Polycystic kidney disease 2 protein ;
Polycystwin ;
R48321
展开内容
背景:
polycystin 2 , transient receptor potential cation channel (PKD2) Homo sapiens This gene encodes a member of the polycystin protein family. The encoded protein is a multi-pass membrane protein that functions as a calcium permeable cation channel , and is involved in calcium transport and calcium signaling in renal epithelial cells. This protein interacts with polycystin 1 , and they may be partners in a common signaling cascade involved in tubular morphogenesis. Mutations in this gene are associated with autosomal dominant polycystic kidney disease type 2. [provided by RefSeq , Mar 2011] ,
展开内容
功能:
Disease:Defects in PKD2 are the cause of polycystic kidney disease autosomal dominant type 2 (ADPKD2) [MIM:173900]. ADPKD2 represents approximately 15% of the cases of ADPKD , a common genetic disease affecting about 1:400 to 1:1000 individuals. ADPKD is characterized by progressive formation and enlargement of cysts in both kidneys , typically leading to end-stage renal disease in adult life. Cysts also occurs in the liver and other organs. ADPKD2 is clinically milder than ADPKD1 but it has a deleterious impact on overall life expectancy. ,Domain:The C-terminal coiled-coil domain binds calcium and undergoes a calcium-induced conformation change. It is implicated in oligomerization and the interaction with PKD1. ,Function:Functions as a calcium permeable cation channel. PKD1 and PKD2 may function through a common signaling pathway that is necessary for normal tubulogenesis. ,online information:Polycystin 2 - Not a C-type lectin ,similarity:Belongs to the polycystin family. ,similarity:Contains 1 EF-hand domain. ,subunit:Forms homooligomers. Interacts with PKD1. PKD1 requires the presence of PKD2 for stable expression. Interacts with CD2AP. ,tissue specificity:Strongly expressed in ovary , fetal and adult kidney , testis , and small intestine. Not detected in peripheral leukocytes. ,
展开内容
细胞定位:
Cell projection , cilium membrane ; Multi-pass membrane protein . Endoplasmic reticulum membrane ; Multi-pass membrane protein . Cell membrane ; Multi-pass membrane protein . Basolateral cell membrane . Cytoplasmic vesicle membrane . Golgi apparatus . PKD2 localization to the plasma and ciliary membranes requires PKD1. PKD1:PKD2 interaction is required to reach the Golgi apparatus form endoplasmic reticulum and then traffic to the cilia (By similarity) . Retained in the endoplasmic reticulum by interaction with PACS1 and PACS2 (PubMed:15692563) . Detected on kidney tubule basolateral membranes and basal cytoplasmic vesicles (PubMed:10770959) . Cell surface and cilium localization requires GANAB (PubMed:27259053) . .
展开内容
文献引用({{totalcount}})
货号: YP1196
规格
价格
货期
数量
200μL
¥4,680.00
现货
0
100μL
¥2,800.00
现货
0
50μL
¥1,500.00
现货
0
加入购物车
已收藏
收藏
Recently Viewed Products
Clear all







×

Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
主要信息
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
产品 {{index}}/{{pcount}}
上一个产品
下一个产品
{{pvTitle}}
滚轮缩放图片
{{pvDescr}}