



VHL (Phospho Ser68) Rabbit pAb
-YP1113


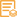


主要信息
Target
VHL Phospho Ser68
Host Species
Rabbit
Reactivity
Human, Mouse, Rat
Applications
IHC, IF, ELISA
MW
19-24kD (Observed)
Conjugate/Modification
Phospho
货号: YP1113
规格
价格
货期
数量
200μL
¥4,680.00
现货
0
100μL
¥2,800.00
现货
0
50μL
¥1,500.00
现货
0
加入购物车


已收藏
收藏
详细信息
推荐稀释比
IHC 1:100-1:300; ELISA 1:5000; IF 1:50-200
组成
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
特异性
Phospho-VHL (S68) Polyclonal Antibody detects endogenous levels of VHL protein only when phosphorylated at S68.The name of modified sites may be influenced by many factors, such as species (the modified site was not originally found in human samples) and the change of protein sequence (the previous protein sequence is incomplete, and the protein sequence may be prolonged with the development of protein sequencing technology). When naming, we will use the "numbers" in historical reference to keep the sites consistent with the reports. The antibody binds to the following modification sequence (lowercase letters are modification sites):VNsRE
纯化工艺
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
储存
-15°C to -25°C/1 year (Do not lower than -25°C)
浓度
1 mg/ml
实测条带
19-24kD
修饰
Phospho
克隆性
Polyclonal
同种型
IgG
相关产品
Primary Antibodies
VHL Rabbit pAb
YT5988
预览→
Primary Antibodies
VHL Rabbit pAb
YT4876
预览→
Primary Antibodies
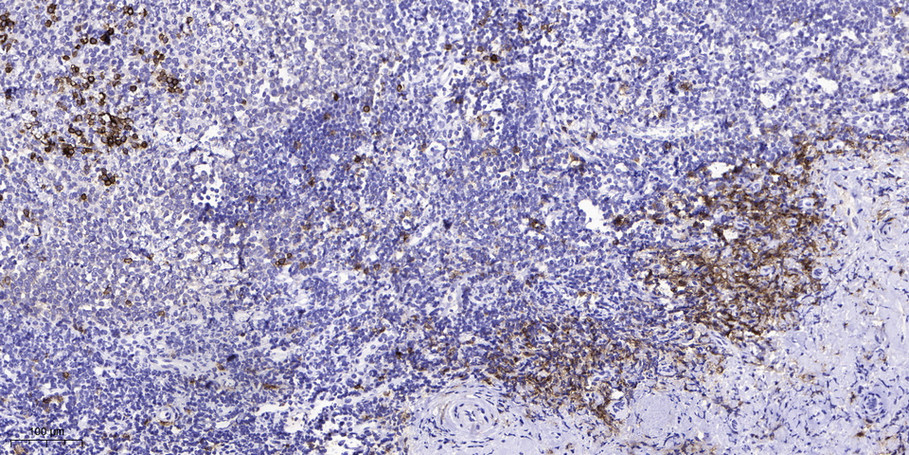
VHL (Phospho Ser68) Rabbit pAb
YP1113
预览→
Primary Antibodies
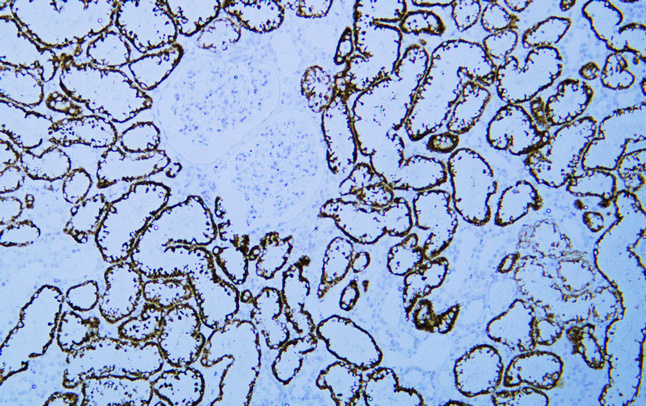
VHL (ABT-PVHL) Mouse mAb
YM6215
预览→
抗原&靶点信息
免疫原:
The antiserum was produced against synthesized peptide derived from human VHL around the phosphorylation site of Ser68. AA range:34-83
展开内容
特异性:
Phospho-VHL (S68) Polyclonal Antibody detects endogenous levels of VHL protein only when phosphorylated at S68.The name of modified sites may be influenced by many factors, such as species (the modified site was not originally found in human samples) and the change of protein sequence (the previous protein sequence is incomplete, and the protein sequence may be prolonged with the development of protein sequencing technology). When naming, we will use the "numbers" in historical reference to keep the sites consistent with the reports. The antibody binds to the following modification sequence (lowercase letters are modification sites):VNsRE
展开内容
基因名称:
VHL
展开内容
蛋白名称:
Von Hippel-Lindau disease tumor suppressor
展开内容
别名:
VHL ;
Von Hippel-Lindau disease tumor suppressor ;
Protein G7 ;
pVHL
Von Hippel-Lindau disease tumor suppressor ;
Protein G7 ;
pVHL
展开内容
背景:
von Hippel-Lindau tumor suppressor (VHL) Homo sapiens Von Hippel-Lindau syndrome (VHL) is a dominantly inherited familial cancer syndrome predisposing to a variety of malignant and benign tumors. A germline mutation of this gene is the basis of familial inheritance of VHL syndrome. The protein encoded by this gene is a component of the protein complex that includes elongin B , elongin C , and cullin-2 , and possesses ubiquitin ligase E3 activity. This protein is involved in the ubiquitination and degradation of hypoxia-inducible-factor (HIF) , which is a transcription factor that plays a central role in the regulation of gene expression by oxygen. RNA polymerase II subunit POLR2G/RPB7 is also reported to be a target of this protein. Alternatively spliced transcript variants encoding distinct isoforms have been observed. [provided by RefSeq , Jul 2008] ,
展开内容
功能:
Disease:Defects in VHL are a cause of pheochromocytoma [MIM:171300]. The pheochromocytomas are catecholamine-producing , chromaffin tumors that arise in the adrenal medulla in 90% of cases. In the remaining 10% of cases , they develop in extra-adrenal sympathetic ganglia and may be referred to as "paraganglioma." Pheochromocytoma usually presents with hypertension. Approximately 10% of pheochromocytoma is hereditary. The genetic basis for most cases of non-syndromic familial pheochromocytoma is unknown. ,Disease:Defects in VHL are a cause of renal cell carcinoma type 1 (RCC1) [MIM:144700]; also called hypernephroma or adenocarcinoma of kidney. Familial renal cell carcinoma syndromes form a group of diseases characterized by a predisposition to development of renal cell carcinomas (RCCs) with various histological subtypes. ,Disease:Defects in VHL are the cause of erythrocytosis familial type 2 (ECYT2) [MIM:263400]; also called VHL-dependent polycythemia or Chuvash type polycythemia. ECYT2 is an autosomal recessive disorder characterized by an increase in serum red blood cell mass , hypersensitivity of erythroid progenitors to erythropoietin , increased erythropoietin serum levels , and normal oxygen affinity. Patients with ECYT2 carry a high risk for peripheral thrombosis and cerebrovascular events. ,Disease:Defects in VHL are the cause of von Hippel-Lindau disease (VHLD) [MIM:193300]. VHLD is a dominantly inherited familial cancer syndrome characterized by the development of retinal angiomatosis , cerebellar and spinal hemangioblastoma , renal cell carcinoma (RCC) , phaeochromocytoma and pancreatic tumors. VHL type 1 is without pheochromocytoma , type 2 is with pheochromocytoma. VHL type 2 is further subdivided into types 2A (pheochromocytoma , retinal angioma , and hemangioblastomas without renal cell carcinoma and pancreatic cyst) and 2B (pheochromocytoma , retinal angioma , and hemangioblastomas with renal cell carcinoma and pancreatic cyst) . VHL type 2C refers to patients with isolated pheochromocytoma without hemangioblastoma or renal cell carcinoma. The estimated incidence is 3/100000 births per year and penetrance is 97% by age 60 years. ,Domain:The elongin BC complex binding domain is also known as BC-box with the consensus [APST]-L-x (3) -C-x (3) -[AILV]. ,Function:Involved in the ubiquitination and subsequent proteasomal degradation via the von Hippel-Lindau ubiquitination complex. Seems to act as target recruitment subunit in the E3 ubiquitin ligase complex and recruits hydroxylated hypoxia-inducible factor (HIF) under normoxic conditions. Involved in transcriptional repression through interaction with HIF1A , HIF1AN and histone deacetylases. ,pathway:Protein modification; protein ubiquitination. ,subcellular location:Equally distributed between the nucleus and the cytoplasm but not membrane-associated. ,subcellular location:Found predominantly in the cytoplasm and with less amounts nuclear or membrane-associated. ,subunit:Component of the VCB (VHL-Elongin BC-CUL2) complex; this complex acts as a ubiquitin-ligase E3 and directs proteosome-dependent degradation of targeted proteins. Interacts with CUL2; this interaction is dependent on the integrity of the trimeric VBC complex. Interacts (via the beta domain) with HIF1A (via the NTAD domain) ; this interaction mediates degradation of HIF1A in normoxia and , in hypoxia , prevents ubiqitination and degradation of HIF1A by mediating hypoxia-induced translocation to the nucleus , a process which requires a hypoxia-dependent regulatory signal. Interacts with RNF139 and UBP33. Interacts with PHF17. ,tissue specificity:Expressed in the adult and fetal brain and kidney. ,
展开内容
细胞定位:
[Isoform 1]: Cytoplasm. Membrane; Peripheral membrane protein. Nucleus. Found predominantly in the cytoplasm and with less amounts nuclear or membrane-associated. Colocalizes with ADRB2 at the cell membrane.; [Isoform 3]: Cytoplasm. Nucleus. Equally distributed between the nucleus and the cytoplasm but not membrane-associated.
展开内容
研究领域:
>>HIF-1 signaling pathway ;
>>Ubiquitin mediated proteolysis ;
>>Pathways in cancer ;
>>Renal cell carcinoma
>>Ubiquitin mediated proteolysis ;
>>Pathways in cancer ;
>>Renal cell carcinoma
展开内容
信号通路
Human Diseases >> Cancer: overview >> Pathways in cancer
Human Diseases >> Cancer: specific types >> Renal cell carcinoma
Environmental Information Processing >> Signal transduction >> HIF-1 signaling pathway
Genetic Information Processing >> Folding, sorting and degradation >> Ubiquitin mediated proteolysis
文献引用({{totalcount}})
货号: YP1113
规格
价格
货期
数量
200μL
¥4,680.00
现货
0
100μL
¥2,800.00
现货
0
50μL
¥1,500.00
现货
0
加入购物车
已收藏
收藏
Recently Viewed Products
Clear all







×

Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
主要信息
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
产品 {{index}}/{{pcount}}
上一个产品
下一个产品
{{pvTitle}}
滚轮缩放图片
{{pvDescr}}