




Flk-1/VEGFR2 (Phospho Tyr1059) Rabbit pAb
-YP0322




主要信息
Target
Flk-1/VEGFR2 Phospho Tyr1059
Host Species
Rabbit
Reactivity
Human, Mouse, Rat
Applications
WB, ELISA
MW
170kD (Observed)
Conjugate/Modification
phosphate
货号: YP0322
规格
价格
货期
数量
200μL
¥4,680.00
现货
0
100μL
¥2,800.00
现货
0
50μL
¥1,500.00
现货
0
加入购物车


已收藏
收藏
详细信息
推荐稀释比
WB 1:500-1:2000; ELISA 1:5000; Not yet tested in other applications.
组成
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
特异性
Phospho-Flk-1 (Y1059) Polyclonal Antibody detects endogenous levels of Flk-1 protein only when phosphorylated at Y1059.The name of modified sites may be influenced by many factors, such as species (the modified site was not originally found in human samples) and the change of protein sequence (the previous protein sequence is incomplete, and the protein sequence may be prolonged with the development of protein sequencing technology). When naming, we will use the "numbers" in historical reference to keep the sites consistent with the reports. The antibody binds to the following modification sequence (lowercase letters are modification sites):PDyVR
纯化工艺
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
储存
-15°C to -25°C/1 year (Do not lower than -25°C)
浓度
1 mg/ml
实测条带
170kD
修饰
phosphate
克隆性
Polyclonal
同种型
IgG
相关产品
Secondary Antibodies
Goat Anti Mouse IgG(H+L) (HRP)
RS0001
预览→
Secondary Antibodies
Goat Anti Rabbit IgG(H+L) (HRP)
RS0002
预览→
Primary Antibodies
β-actin (PTR2364) Mouse mAb
YM3028
预览→
Primary Antibodies
GAPDH (PTR2304) Mouse mAb
YM3029
预览→
抗原&靶点信息
免疫原:
The antiserum was produced against synthesized peptide derived from human VEGFR2 around the phosphorylation site of Tyr1059. AA range:1025-1074
展开内容
特异性:
Phospho-Flk-1 (Y1059) Polyclonal Antibody detects endogenous levels of Flk-1 protein only when phosphorylated at Y1059.The name of modified sites may be influenced by many factors, such as species (the modified site was not originally found in human samples) and the change of protein sequence (the previous protein sequence is incomplete, and the protein sequence may be prolonged with the development of protein sequencing technology). When naming, we will use the "numbers" in historical reference to keep the sites consistent with the reports. The antibody binds to the following modification sequence (lowercase letters are modification sites):PDyVR
展开内容
基因名称:
KDR FLK1 VEGFR2
展开内容
蛋白名称:
Vascular endothelial growth factor receptor 2
展开内容
别名:
KDR ;
FLK1 ;
VEGFR2 ;
Vascular endothelial growth factor receptor 2 ;
VEGFR-2 ;
Fetal liver kinase 1 ;
FLK-1 ;
Kinase insert domain receptor ;
KDR ;
Protein-tyrosine kinase receptor flk-1 ;
CD antigen CD309
FLK1 ;
VEGFR2 ;
Vascular endothelial growth factor receptor 2 ;
VEGFR-2 ;
Fetal liver kinase 1 ;
FLK-1 ;
Kinase insert domain receptor ;
KDR ;
Protein-tyrosine kinase receptor flk-1 ;
CD antigen CD309
展开内容
背景:
Vascular endothelial growth factor (VEGF) is a major growth factor for endothelial cells. This gene encodes one of the two receptors of the VEGF. This receptor , known as kinase insert domain receptor , is a type III receptor tyrosine kinase. It functions as the main mediator of VEGF-induced endothelial proliferation , survival , migration , tubular morphogenesis and sprouting. The signalling and trafficking of this receptor are regulated by multiple factors , including Rab GTPase , P2Y purine nucleotide receptor , integrin alphaVbeta3 , T-cell protein tyrosine phosphatase , etc.. Mutations of this gene are implicated in infantile capillary hemangiomas. [provided by RefSeq , May 2009] ,
展开内容
功能:
Catalytic activity:ATP + a [protein]-L-tyrosine = ADP + a [protein]-L-tyrosine phosphate. ,Function:Receptor for VEGF or VEGFC. Has a tyrosine-protein kinase activity. The VEGF-kinase ligand/receptor signaling system plays a key role in vascular development and regulation of vascular permeability. In case of HIV-1 infection , the interaction with extracellular viral Tat protein seems to enhance angiogenesis in Kaposi's sarcoma lesions. ,similarity:Belongs to the protein kinase superfamily. Tyr protein kinase family. ,similarity:Belongs to the protein kinase superfamily. Tyr protein kinase family. CSF-1/PDGF receptor subfamily. ,similarity:Contains 1 protein kinase domain. ,similarity:Contains 7 Ig-like C2-type (immunoglobulin-like) domains. ,subunit:Interacts with MYOF (By similarity) . Interacts with SHB; upon VEGF activation. Interacts with HIV-1 Tat. ,
展开内容
细胞定位:
Cell junction . Endoplasmic reticulum . Cell membrane . Localized with RAP1A at cell-cell junctions (By similarity) . Colocalizes with ERN1 and XBP1 in the endoplasmic reticulum in endothelial cells in a vascular endothelial growth factor (VEGF) -dependent manner (PubMed:23529610) . .; [Isoform 1]: Cell membrane; Single-pass type I membrane protein. Cytoplasm. Nucleus. Cytoplasmic vesicle. Early endosome. Detected on caveolae-enriched lipid rafts at the cell surface. Is recycled from the plasma membrane to endosomes and back again. Phosphorylation triggered by VEGFA binding promotes internalization and subsequent degradation. VEGFA binding triggers internalization and translocation to the nucleus.; [Isoform 2]: Secreted .; [Isoform 3]: Secreted.
展开内容
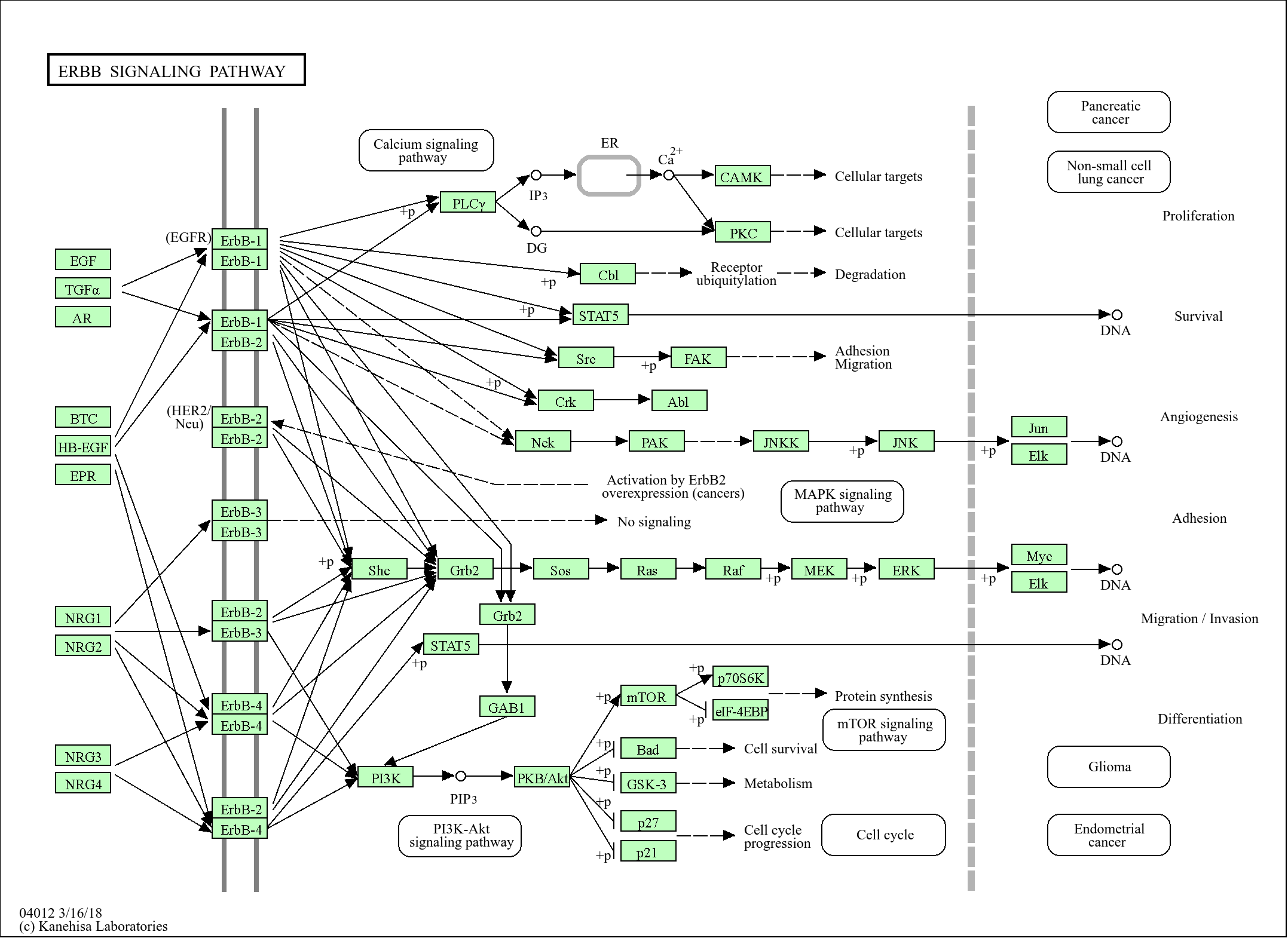
研究领域:
>>EGFR tyrosine kinase inhibitor resistance ;
>>MAPK signaling pathway ;
>>Ras signaling pathway ;
>>Rap1 signaling pathway ;
>>Calcium signaling pathway ;
>>PI3K-Akt signaling pathway ;
>>VEGF signaling pathway ;
>>Focal adhesion ;
>>Proteoglycans in cancer ;
>>Fluid shear stress and atherosclerosis
>>MAPK signaling pathway ;
>>Ras signaling pathway ;
>>Rap1 signaling pathway ;
>>Calcium signaling pathway ;
>>PI3K-Akt signaling pathway ;
>>VEGF signaling pathway ;
>>Focal adhesion ;
>>Proteoglycans in cancer ;
>>Fluid shear stress and atherosclerosis
展开内容
信号通路
Cellular Processes >> Cellular community - eukaryotes >> Focal adhesion
Environmental Information Processing >> Signal transduction >> MAPK signaling pathway
Environmental Information Processing >> Signal transduction >> Ras signaling pathway
Environmental Information Processing >> Signal transduction >> Rap1 signaling pathway
Environmental Information Processing >> Signal transduction >> VEGF signaling pathway
Environmental Information Processing >> Signal transduction >> Calcium signaling pathway
Environmental Information Processing >> Signal transduction >> PI3K-Akt signaling pathway
文献引用({{totalcount}})
货号: YP0322
规格
价格
货期
数量
200μL
¥4,680.00
现货
0
100μL
¥2,800.00
现货
0
50μL
¥1,500.00
现货
0
加入购物车
已收藏
收藏
Recently Viewed Products
Clear all







×

Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
主要信息
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
产品 {{index}}/{{pcount}}
上一个产品
下一个产品
{{pvTitle}}
滚轮缩放图片
{{pvDescr}}