



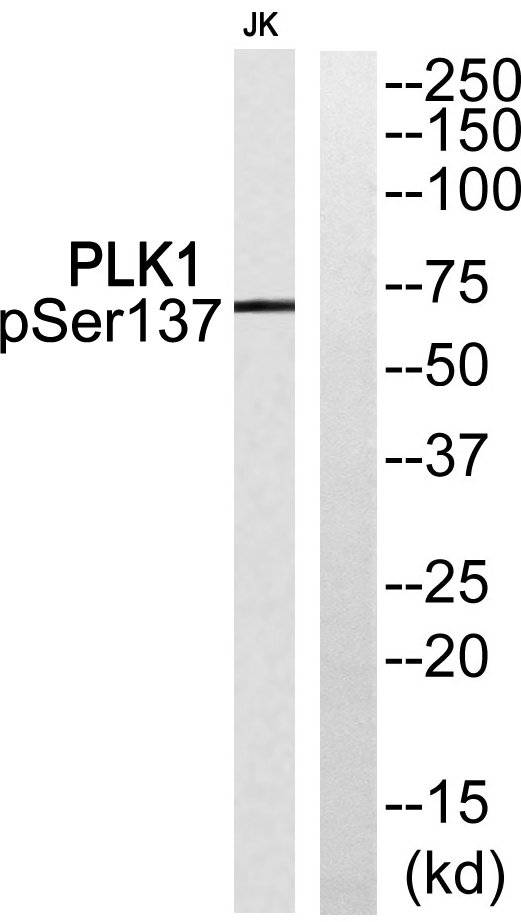
PLK1 (Phospho Ser137) Rabbit pAb
-YP0434




主要信息
Target
PLK1 Phospho Ser137
Host Species
Rabbit
Reactivity
Human, Mouse, Rat
Applications
WB, IF, ELISA, IHC
MW
68kD (Observed)
Conjugate/Modification
Phospho
货号: YP0434
规格
价格
货期
数量
200μL
¥4,680.00
现货
0
100μL
¥2,800.00
现货
0
50μL
¥1,500.00
现货
0
加入购物车


已收藏
收藏
详细信息
推荐稀释比
WB 1:500-2000; IF/ICC1:50-200; ELISA 1:2000-20000; IHC 1:50-200
组成
Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide.
特异性
Phospho-Plk1 (S137) Polyclonal Antibody detects endogenous levels of Plk1 protein only when phosphorylated at S137.The name of modified sites may be influenced by many factors, such as species (the modified site was not originally found in human samples) and the change of protein sequence (the previous protein sequence is incomplete, and the protein sequence may be prolonged with the development of protein sequencing technology). When naming, we will use the "numbers" in historical reference to keep the sites consistent with the reports. The antibody binds to the following modification sequence (lowercase letters are modification sites):RRsLL
纯化工艺
The antibody was affinity-purified from rabbit antiserum by affinity-chromatography using epitope-specific immunogen.
储存
-15°C to -25°C/1 year (Do not lower than -25°C)
浓度
1 mg/ml
实测条带
68kD
修饰
Phospho
克隆性
Polyclonal
同种型
IgG
相关产品
Primary Antibodies

PLK1 Rabbit pAb
YT5054
预览→
Primary Antibodies
PLK1 Rabbit pAb
YT3797
预览→
Primary Antibodies
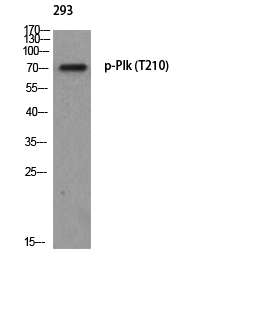
PLK1 (Phospho Thr210) Rabbit pAb
YP0964
预览→
Primary Antibodies
PLK1 (Phospho Ser137) Rabbit pAb
YP0434
预览→
ELISA Kits
Total PLK1 Cell-Based Colorimetric ELISA Kit
KA4248C
预览→
ELISA Kits
PLK1 (Phospho Ser137) Cell-Based Colorimetric ELISA Kit
KA1720C
预览→
抗原&靶点信息
免疫原:
The antiserum was produced against synthesized peptide derived from human PLK1 around the phosphorylation site of Ser137. AA range:103-152
展开内容
特异性:
Phospho-Plk1 (S137) Polyclonal Antibody detects endogenous levels of Plk1 protein only when phosphorylated at S137.The name of modified sites may be influenced by many factors, such as species (the modified site was not originally found in human samples) and the change of protein sequence (the previous protein sequence is incomplete, and the protein sequence may be prolonged with the development of protein sequencing technology). When naming, we will use the "numbers" in historical reference to keep the sites consistent with the reports. The antibody binds to the following modification sequence (lowercase letters are modification sites):RRsLL
展开内容
基因名称:
PLK1
展开内容
蛋白名称:
Serine/threonine-protein kinase PLK1
展开内容
别名:
PLK1 ;
PLK ;
Serine/threonine-protein kinase PLK1 ;
Polo-like kinase 1 ;
PLK-1 ;
Serine/threonine-protein kinase 13 ;
STPK13
PLK ;
Serine/threonine-protein kinase PLK1 ;
Polo-like kinase 1 ;
PLK-1 ;
Serine/threonine-protein kinase 13 ;
STPK13
展开内容
背景:
The Ser/Thr protein kinase encoded by this gene belongs to the CDC5/Polo subfamily. It is highly expressed during mitosis and elevated levels are found in many different types of cancer. Depletion of this protein in cancer cells dramatically inhibited cell proliferation and induced apoptosis; hence , it is a target for cancer therapy. [provided by RefSeq , Sep 2015] ,
展开内容
功能:
Catalytic activity:ATP + a protein = ADP + a phosphoprotein. ,developmental stage:Accumulates to a maximum during the G2 and M phases , declines to a nearly undetectable level following mitosis and throughout G1 phase , and then begins to accumulate again during S phase. ,enzyme regulation:Activated by serine and threonine phosphorylation. ,Function:Serine/threonine-protein kinase that performs several important functions throughout M phase of the cell cycle , including the regulation of centrosome maturation and spindle assembly , the removal of cohesins from chromosome arms , the inactivation of APC/C inhibitors , and the regulation of mitotic exit and cytokinesis. ,induction:By growth-stimulating agents. ,PTM:Autophosphorylation and phosphorylation of Ser-137 are not significant events during activation of PLK1 in M phase. ,PTM:Catalytic activity is enhanced by phosphorylation of Thr-210 and/or Ser-137. ,similarity:Belongs to the protein kinase superfamily. ,similarity:Belongs to the protein kinase superfamily. Ser/Thr protein kinase family. CDC5/Polo subfamily. ,similarity:Contains 1 protein kinase domain. ,similarity:Contains 2 POLO box domains. ,subunit:Interacts with CEP170 and EVI5. Interacts and phosphorylates ERCC6L. Interacts with FAM29A. ,tissue specificity:Placenta and colon. ,
展开内容
细胞定位:
Nucleus. Chromosome , centromere , kinetochore. Cytoplasm , cytoskeleton , microtubule organizing center , centrosome . Cytoplasm , cytoskeleton , spindle . Midbody . localization at the centrosome starts at the G1/S transition (PubMed:24018379) . During early stages of mitosis , the phosphorylated form is detected on centrosomes and kinetochores. Localizes to the outer kinetochore. Presence of SGO1 and interaction with the phosphorylated form of BUB1 is required for the kinetochore localization. Localizes onto the central spindle by phosphorylating and docking at midzone proteins KIF20A/MKLP2 and PRC1. Colocalizes with FRY to separating centrosomes and spindle poles from prophase to metaphase in mitosis , but not in other stages of the cell cycle. Localization to the centrosome is required for S phase progression (PubMed:24018379) . Colocalizes with HSF1 at the spindle poles during prometaphase (PubMed:18794143) . .
展开内容
组织表达:
展开内容
研究领域:
>>FoxO signaling pathway ;
>>Cell cycle ;
>>Oocyte meiosis ;
>>Progesterone-mediated oocyte maturation
>>Cell cycle ;
>>Oocyte meiosis ;
>>Progesterone-mediated oocyte maturation
展开内容
信号通路
文献引用({{totalcount}})
货号: YP0434
规格
价格
货期
数量
200μL
¥4,680.00
现货
0
100μL
¥2,800.00
现货
0
50μL
¥1,500.00
现货
0
加入购物车
已收藏
收藏
Recently Viewed Products
Clear all







×

Toggle night Mode
{{pinfoXq.title || ''}}
Catalog: {{pinfoXq.catalog || ''}}
Filter:
All
{{item.name}}
{{pinfo.title}}
-{{pinfo.catalog}}
主要信息
Target
{{pinfo.target}}
Reactivity
{{pinfo.react}}
Applications
{{pinfo.applicat}}
Conjugate/Modification
{{pinfo.coupling}}/{{pinfo.modific}}
MW (kDa)
{{pinfo.mwcalc}}
Host Species
{{pinfo.hostspec}}
Isotype
{{pinfo.isotype}}
产品 {{index}}/{{pcount}}
上一个产品
下一个产品
{{pvTitle}}
滚轮缩放图片
{{pvDescr}}